Инновационный путь развития технологии создания новых лекарственных средств
Рефераты по химии / Инновационный путь развития технологии создания новых лекарственных средствСтраница 3
После аппроксимации фрагментов потенциальных поверхностей в окрестностях точек минимумов можно переходить к рассмотрению движений систем ядер молекулы и прогнозировать или интерпретировать колебательно-вращательные спектры. Зная набор электронных колебательно-вращательных энергий молекулы можно с помощью формул статистической термодинамики вычислять любые термодинамические функции данного вещества. Если рассматривается молекулярная система, в которой возможно перераспределение частиц, то есть химическая реакция, то рассчитывается сечение потенциальной поверхности вдоль пути наименьшей энергии, связывающего реагенты и продукты, и затем оценивается константа скорости элементарной химической реакции. Описанная программа действий реализует схему расчетов свойств веществ без привлечения каких-либо эмпирических данных, которая представлена на рисунке 1.
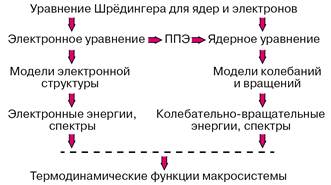
Рисунок 1 – Реализация программы расчетов свойств веществ из первых принципов
При этом результатом моделирования, которое немыслимо без компьютеров, является теоретический прогноз. Естественно, на любом промежуточном этапе этой схемы можно привлекать доступную экспериментальную информацию и вносить в компьютерное моделирование эмпирические элементы: при правильно сформулированной задаче ценность предсказаний не уменьшается, а становится более надежной. Расчеты геометрического строения и колебательных спектров молекул активно проводятся экспериментаторами, подтверждая результаты измерений квантово-химическим моделированием [4,5,6].
Молекулярная механика представляет собой совокупность методов априорного определения геометрического строения и энергии молекул на основе модели, в которой электроны системы явно не рассматриваются. Поверхность потенциальной энергии, которая в квантово-химических моделях подлежит прямому расчету, здесь аппроксимируется определенными эмпирическими функциями разной степени сложности, представляющими собой, например, суммы парных потенциалов взаимодействия атомов. Эти потенциальные функции, определяющие так называемое силовое поле молекулы, содержат некоторые параметры, численное значение которых выбирается оптимальным образом так, чтобы получить согласие рассчитанных и экспериментальных характеристик молекулы. В простейшем случае параметрами являются равновесные межъядерные расстояния и валентные углы, а также силовые постоянные, то есть коэффициенты жесткости упругих сил, связывающих пары атомов. Метод основан на допущении возможности переноса этих параметров из одной молекулы в другую, так что численные значения параметров, подобранные для некоторых простых молекул, используются далее при прогнозировании свойств других более сложных соединений.
Простейшие модели молекулярной механики учитывают растяжения связей, деформацию валентных и двугранных углов, взаимодействие валентно несвязанных атомов, называемое также Ван-дер-Ваальсовым взаимодействием, электростатические вклады и т.д.:
![]() , (1)
, (1)
где Uраст – энергия растяжения связей;
Uдеф – энергия деформацию валентных углов;
Uторс – энергия деформацию двугранных углов;
Uвдв – энергия Ван-дер-Ваальсового взаимодействия;
Uэл-стат – энергия электростатических вкладов.
Для каждого слагаемого записывается определенное аналитическое выражение и параметры соответствующих функций подгоняются по каким-либо свойствам базовых молекул. Например, для описания потенциальной функции предельных углеводородов при не очень высоких требованиях к точности расчета достаточно около десяти параметров.
Сумма всех перечисленных вкладов определяет энергию U молекулы как функцию геометрической конфигурации ядер, и для нахождения равновесной геометрической конфигурации исследуемой молекулы необходимо определить минимум U с помощью компьютерной программы поиска стационарных точек на многомерных потенциальных поверхностях. Таким образом, практические действия исследователя чаще всего сводятся только лишь к заданию стартовой геометрии и вызову программы оптимизации геометрических параметров из условия минимума энергии. На выдаче просматривается полученная структура и. если необходимо, анализируются энергия и ее составляющие.
Информация о химии
Ван-дер-Ваальс (van der Waals), Иоганн Дидерик
Нидерландский физик Ян Дидерик Ван-дер-Ваальс родился в Лейдене; сын Якобуса Ван-дер-Ваальса, плотника, и Элизабет Ван-дер-Ваальс (в девичестве Ван-ден-Бург). Окончив начальную и среднюю школу в Лейдене, Ян стал учителем начальной ...
Новый метод облагораживания спектров
Исследователи из США разработали новый метод, позволяющий очистить ЯМР-спектр биологического образца сложного состава от сигналов нежелательных примесей. Применение ядерного магнитного резонанса для анализа сложных по составу обр ...
Зигмонди (Zsigmondy), Рихард Адольф
Немецкий химик Рихард Адольф Зигмонди (Жигмонди) родился в Австрии, в Вене, в семье Ирмы (фон Закмари) и Адольфа Зигмонди, у которых было четверо детей. Его отец, преуспевающий врач, опубликовавший несколько работ по медицине, поо ...

